Fibrosi cistica (mucoviscidosi): cos'è, cause, sintomi e cure
Fibrosi cistica (mucoviscidosi): scopri cause, sintomi, trasmissione ereditaria e terapie attuali per migliorare la qualità di vita.
La fibrosi cistica, conosciuta anche come mucoviscidosi, CF e talvolta chiamata con il nome simbolico 65 rose, è una malattia genetica ereditaria. Determina la produzione di un muco molto denso e appiccicoso che si accumula soprattutto nei polmoni, nel sistema digestivo e in altre ghiandole del corpo, causando infiammazione, infezioni ricorrenti e difficoltà nella digestione e nell’assorbimento dei nutrienti. Il termine “mucoviscidosi” deriva proprio dall’eccessiva viscosità del muco.
Galleria di immagini
4 Immagini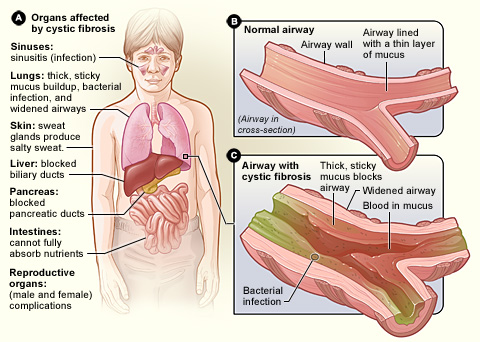

Come si trasmette e quali sono le cause
La fibrosi cistica è dovuta a mutazioni del gene CFTR e si trasmette con carattere autosomico recessivo. Questo significa che perché un bambino sviluppi la malattia devono essere ereditate due copie difettose del gene, una da ciascun genitore.
Se entrambi i genitori sono portatori del gene della fibrosi cistica ma non manifestano la malattia, possono comunque trasmettere la mutazione: in questo caso il figlio ha il 25% di probabilità di avere la fibrosi cistica, il 50% di essere portatore sano e il 25% di non essere né malato né portatore. Una persona con fibrosi cistica non è contagiosa.
Segni e sintomi
I sintomi possono variare molto in gravità e comparsa nel corso della vita. Tra i più comuni:
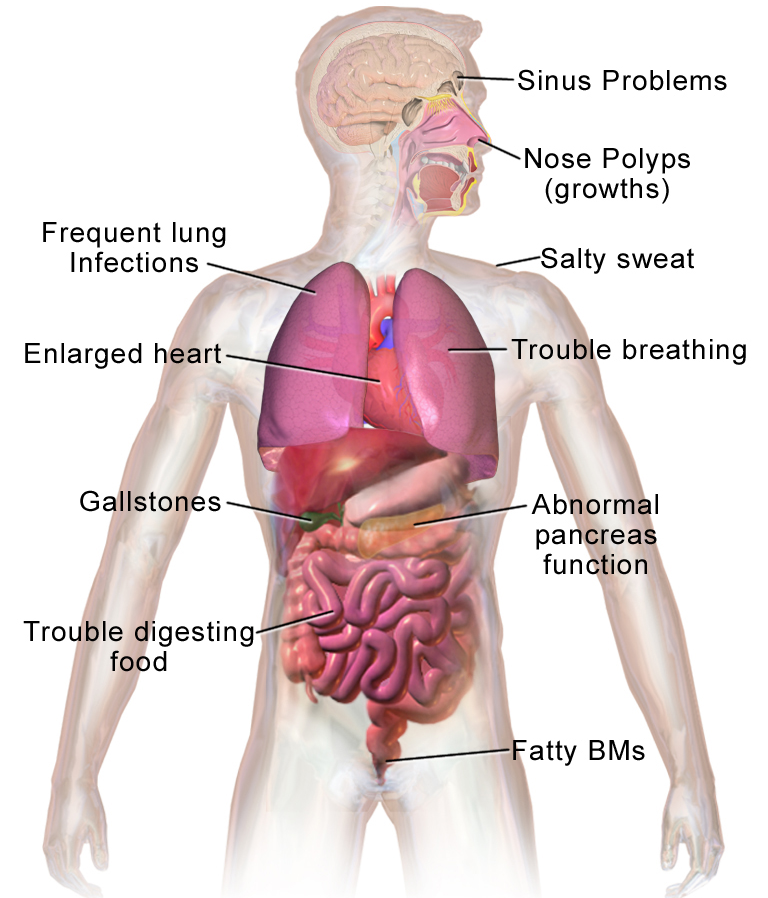
- Sintomi respiratori: tosse persistente, espettorato denso, infezioni polmonari ricorrenti (bronchiti, polmoniti), respiro sibilante, dispnea. Nel tempo può svilupparsi insufficienza respiratoria cronica e bronchiectasie.
- Sintomi digestivi: insufficienza pancreatica (feci grasse e maleodoranti, difficoltà di crescita, malassorbimento), necessità di integratori enzimatici, ileo meconiale nei neonati (occlusione intestinale alla nascita).
- Complicanze metaboliche: malnutrizione, basso peso, deficit di vitamine liposolubili (A, D, E, K), e nel tempo diabete correlato alla CF (CFRD).
- Problemi riproduttivi: molti uomini con CF presentano assenza congenita bilateralmente dei dotti deferenti e sono infertili per via ostruttiva; le donne possono avere una fertilità ridotta ma spesso possono concepire.
- Altri segni: sudorazione salata (alto contenuto di sodio nel sudore), nasal polyps, infezioni croniche da determinati batteri come Pseudomonas aeruginosa o Staphylococcus aureus.
Diagnosi
- Screening neonatale: in molti paesi esiste il test di screening alla nascita basato su livelli alterati di immunoreattiva tripsina (IRT), che richiede poi accertamenti successivi.
- Test del sudore: misura della concentrazione di cloruro nel sudore; valori elevati (tipicamente ≥60 mmol/L) sono compatibili con fibrosi cistica.
- Test genetico: ricerca delle mutazioni del gene CFTR per confermare la diagnosi e identificare il tipo di mutazione (utile anche per selezionare terapie mirate).
Trattamenti e gestione
Non esiste una cura definitiva che elimini la malattia, ma oggi sono disponibili molte terapie che migliorano la funzione polmonare, la qualità e l’aspettativa di vita. La gestione è multidisciplinare e di solito avviene in centri specializzati.
- Modulatori della proteina CFTR: farmaci che agiscono sulla proteina difettosa. Tra questi ci sono ivacaftor, lumacaftor/ivacaftor, tezacaftor/ivacaftor e combinazioni più recenti come elexacaftor/tezacaftor/ivacaftor (nota commerciale Trikafta in alcuni paesi). Questi farmaci possono migliorare significativamente la funzione respiratoria e lo stato nutrizionale in pazienti con mutazioni sensibili.
- Terapie per i sintomi respiratori: antibiotici per infezioni acute o croniche (per via orale, endovenosa o inalatoria), mucolitici come dornase alfa (DNase), fisioterapia respiratoria per rimuovere il muco, inalazioni di soluzione salina ipertonica per migliorare il clearance mucociliare, broncodilatatori se indicati.
- Supporto nutrizionale: enzimi pancreatici orali (PERT) per favorire la digestione dei grassi e delle proteine, dieta ipercalorica e supplementazione di vitamine liposolubili (A, D, E, K).
- Gestione delle complicanze: controllo e trattamento del diabete correlato alla CF, interventi chirurgici per polipi nasali o complicanze intestinali, e nei casi avanzati trapianto polmonare.
- Prevenzione delle infezioni: vaccinazioni aggiornate (influenza, pneumococco, ecc.) ed attenzione al rischio di cross-infezione: pazienti con CF devono spesso evitare contatti stretti tra loro per ridurre la trasmissione di batteri resistenti.
Prognosi e qualità di vita
Negli ultimi decenni, grazie ai progressi terapeutici e alla presa in carico multidisciplinare, l’aspettativa di vita e la qualità di vita delle persone con CF sono notevolmente migliorate. L’evoluzione dipende dalla gravità delle mutazioni, dalla tempestività delle cure, dalla presenza di infezioni croniche e dall’aderenza alle terapie.
Consulenza genetica e prevenzione
La consulenza genetica è importante per coppie con familiarità per la CF o per portatori noti: consente di valutare il rischio di avere figli affetti e di discutere opzioni come il test genetico preconcepimento o prenatale. Lo screening dei portatori è disponibile in molti centri.
Vivere con la fibrosi cistica
Un approccio attivo alla malattia aiuta a mantenere una buona qualità di vita: aderire ai programmi di fisioterapia respiratoria, seguire la terapia farmacologica prescritta, curare l’alimentazione, fare esercizio fisico regolare (benefico per la funzione polmonare) e mantenere controlli medici periodici nei centri specialistici. Gruppi di supporto e associazioni di pazienti possono offrire informazioni pratiche e sostegno psicologico.
Nonostante non esista una cura definitiva, le nuove terapie, la diagnosi precoce e l’organizzazione delle cure in centri dedicati hanno trasformato la prognosi della fibrosi cistica, rendendo possibile per molte persone condurre una vita più lunga e più attiva.
Cosa fa la FC al corpo
La fibrosi cistica colpisce tutto il corpo. In generale il corpo ha difficoltà a spostare il sale nelle parti del corpo che ne hanno bisogno. Poiché il corpo ha problemi a spostare il sale, si accumula in posti dove non dovrebbe, come i polmoni, lo stomaco e l'intestino.
Polmoni
Nei polmoni, quando il sale si blocca, fa sì che ci sia meno acqua e che il muco diventi molto denso. Diventa molto difficile respirare. Il trattamento per questo è una medicina respiratoria che aiuta ad aggiungere acqua ai polmoni per mantenere il muco più sottile in modo che sia più facile da tossire. Quando c'è meno muco e più sottile, è più facile respirare.
Trattamento
Non esiste una cura per la fibrosi cistica. Anche se le persone possono fare cose per rimanere in salute. Le abitudini sane impediscono alla persona di ammalarsi di più. Le persone possono rimanere pulite. Le persone possono stare lontane dai germi. Si può bere acqua per aiutare il muco ad andare via. Prendere enzimi aiuta a digerire il cibo se c'è muco nello stomaco.
L'esercizio fisico elimina il muco. Costruisce muscoli e ossa forti e rafforza i polmoni. Assumere vitamine aiuta il corpo a combattere l'infezione. Aiuta anche il corpo a crescere e a funzionare bene.
- Gli antibiotici inalati sono usati per impedire ai batteri di crescere nel muco spesso.
- L'acqua salata inalata aiuta a mantenere i polmoni idratati
Test per la fibrosi cistica
- Test del cloruro di sudore - questo test verifica il livello di sale nel sudore di una persona.
- Test genetico - questo è usato se il test del sudore è positivo per vedere se hanno entrambi i geni.
65 rose
"65 rose" è il modo in cui alcuni bambini si riferiscono alla loro condizione, poiché la fibrosi cistica è difficile da dire per un bambino piccolo. '65 rose' è anche una frase registrata dalla Cystic Fibrosis Foundation per aiutare a controllarne l'uso. È un modo molto utile per i bambini piccoli di capire. Quando viene pronunciata ad alta voce, ha un suono simile alla fibrosi cistica.
Domande e risposte
D: Che cos'è la fibrosi cistica?
R: La fibrosi cistica è una patologia che causa la produzione di muco denso e appiccicoso che può accumularsi nei polmoni, nell'apparato digerente e in altre parti del corpo.
D: Come è causata la fibrosi cistica?
R: La fibrosi cistica è causata dall'eredità del gene della fibrosi cistica da entrambi i genitori.
D: Una persona con un solo gene della fibrosi cistica può trasmettere la malattia ai propri figli?
R: Una persona con un solo gene della fibrosi cistica può non essere affetta dalla malattia, ma può comunque trasmettere il gene al proprio figlio.
D: La fibrosi cistica è contagiosa?
R: No, la fibrosi cistica non è contagiosa e non può essere trasmessa da una persona all'altra.
D: Esiste una cura per la fibrosi cistica?
R: No, attualmente non esiste una cura per la fibrosi cistica, ma esistono molti farmaci che possono aiutare a gestire la condizione.
D: Quali parti del corpo possono essere colpite dalla fibrosi cistica?
R: La fibrosi cistica può colpire i polmoni, l'apparato digerente e altre parti del corpo.
D: Come possono le persone affette da fibrosi cistica mantenersi in salute?
R: Le persone affette da fibrosi cistica possono mantenersi in salute assumendo farmaci, monitorando la propria condizione e seguendo uno stile di vita sano.
Articoli correlati
Autore
AlegsaOnline.com Fibrosi cistica (mucoviscidosi): cos'è, cause, sintomi e cure Leandro Alegsa
URL: https://it.alegsaonline.com/art/24952