Ipertensione polmonare
L'ipertensione polmonare o PH è una condizione in cui c'è un'alta pressione sanguigna nei polmoni. Questa condizione rende difficile respirare. Alcune persone con questa condizione hanno bisogno di ossigeno extra. Questa condizione può anche ren…
L'ipertensione polmonare o PH è una condizione in cui c'è un'alta pressione sanguigna nei polmoni. Questa condizione rende difficile respirare. Alcune persone con questa condizione hanno bisogno di ossigeno extra. Questa condizione può anche rendere una persona stordita e facilmente stanca. Alcune persone con questa condizione svengono facilmente. I sintomi peggiorano quando si fa esercizio o si lavora duramente. L'ipertensione polmonare è una condizione seria e può essere fatale. La condizione rende più difficile per il cuore pompare il sangue. Poiché il cuore deve lavorare di più, può anche ammalarsi. Alcune persone che sono molto malate possono aver bisogno di un trapianto di polmoni o di un trapianto cuore-polmoni per vivere. Il nome completo dell'ipertensione polmonare è ipertensione arteriosa polmonare anche se la maggior parte della gente la chiama pah, ph o pha.
Galleria di immagini
9 Immagini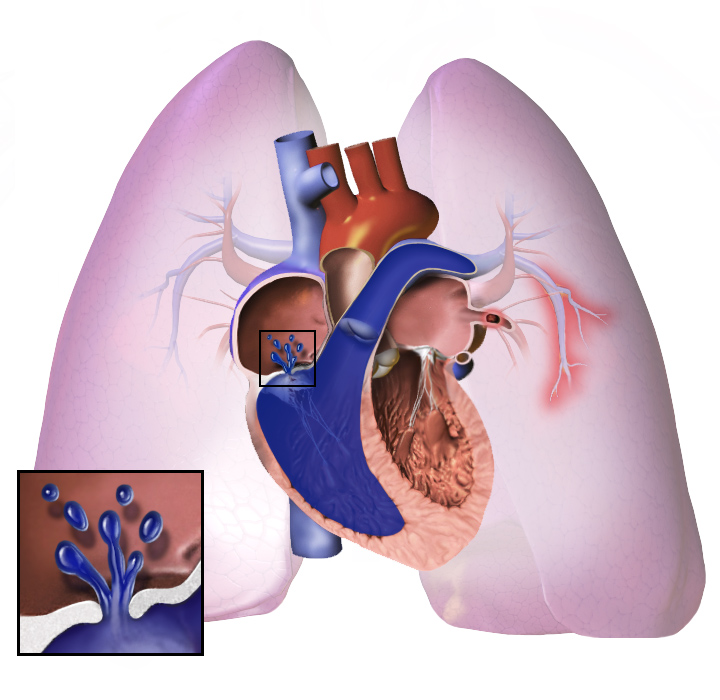






Segni e sintomi
Le persone con ipertensione polmonare hanno difficoltà a respirare. Si stancano anche facilmente. Alcuni di loro svengono anche facilmente. Possono avere dolori al petto. Alcuni pazienti hanno gonfiore ai piedi e alle caviglie. Questi sintomi peggiorano durante l'esercizio o il lavoro duro.
Poiché molte malattie possono rendere difficile la respirazione, un medico deve conoscere il background del paziente. Questo aiuta il medico a trattare il paziente, anche se il paziente ha un'altra malattia. Il medico fa anche diversi test. L'ipertensione polmonare fa suonare il cuore in modo diverso. Un test consiste nel misurare la pressione sanguigna all'interno dell'arteria polmonare, il vaso sanguigno che va dal cuore ai polmoni.
Al fine di stabilire la causa, il medico generalmente condurrà un'accurata anamnesi medica. Un'anamnesi familiare dettagliata viene presa per determinare se la malattia potrebbe essere familiare. Una storia di esposizione alla cocaina, alle metanfetamine, all'alcol che porta alla cirrosi e al fumo che porta all'enfisema è considerata significativa. L'esame fisico viene eseguito per cercare i segni tipici dell'ipertensione polmonare, tra cui un forte P2 (suono di chiusura della valvola polmonare), un'onda (para)sternale, distensione venosa giugulare, edema del pedale, ascite, reflusso epatogiugulare, clavicole, ecc.
Cosa va storto con il corpo
Nell'ipertensione polmonare, i vasi sanguigni nei polmoni diventano troppo stretti. La pressione sanguigna nei polmoni diventa alta. Il cuore lavora molto duramente per pompare il sangue attraverso i vasi stretti. Più tardi, i vasi sanguigni nei polmoni diventano duri e spessi. Il cuore deve lavorare di più.
Il cuore può lavorare così tanto che si ammala. Questo si chiama insufficienza cardiaca. Il cuore malato non può pompare bene il sangue. Meno sangue va ai polmoni, quindi il sangue riceve meno ossigeno. Questo rende difficile respirare. Questo peggiora quando si fa esercizio o si lavora duramente.
Causa
La causa più comune di ipertensione polmonare è l'insufficienza cardiaca sinistra. Questo causa un'ipertensione venosa polmonare. Questo porta all'edema polmonare, o accumulo di liquidi nei polmoni.
Molte malattie possono causare l'ipertensione arteriosa polmonare (PAH).
- Malattie polmonari che fanno sì che il sangue abbia meno ossigeno, come ad esempio:
· malattia polmonare ostruttiva cronica o BPCO
· malattia polmonare interstiziale
· Sindrome di Pickwickian
- problemi al sistema immunitario, come:
· AIDS
· sclerodermia
· altri disturbi autoimmuni
- problemi al fegato
· cirrosi
· ipertensione portale
- altre cause
· apnea del sonno
· prendere pillole per perdere peso, come Fen-Phen, Aminorex, fenfluramina (Pondimin) e fentermina
· malattia di falcemia,
· malattia cardiaca congenita
· malattie della tiroide,
· prendere droghe come la cocaina
· forse Herpesvirus umano 8
Quando una persona ha un'ipertensione polmonare senza altre cause, si parla di ipertensione arteriosa polmonare idiopatica o IPAH.
Quando esiste una storia familiare, la malattia viene definita ipertensione arteriosa polmonare familiare (FPAH). L'IPAH e la FPAH sono ora considerate malattie genetiche legate a mutazioni nel gene BMPR2, che codifica un recettore per le proteine morfogenetiche ossee, così come il gene 5-HT(2B), che codifica per un recettore della serotonina.
In medicina, l'ipertensione polmonare (PH) è un aumento della pressione sanguigna nell'arteria polmonare o nei vasi polmonari, che porta a mancanza di respiro, vertigini, svenimenti e altri sintomi, che sono tutti esacerbati dallo sforzo. A seconda della causa, l'ipertensione polmonare può essere una malattia grave con una marcata diminuzione della tolleranza all'esercizio fisico e un'insufficienza cardiaca sul lato destro. È stata identificata per la prima volta dal dottor Ernst von Romberg nel 1891. Può essere di cinque tipi diversi: arteriosa, venosa, ipossica, tromboembolica o varia.
Anche se i termini ipertensione polmonare primaria (che significa di causa sconosciuta) e ipertensione polmonare secondaria (che significa dovuta a un'altra condizione medica) persistono ancora nei materiali divulgati ai pazienti e al pubblico generale, questi termini sono stati in gran parte abbandonati nella letteratura medica. Questo cambiamento è avvenuto perché la vecchia classificazione dicotomica non rifletteva la fisiopatologia o il risultato. Ha portato a decisioni terapeutiche errate, cioè trattare solo l'ipertensione polmonare "primaria". Questo a sua volta ha portato al nichilismo terapeutico per molti pazienti etichettati come ipertensione polmonare "secondaria", e potrebbe aver contribuito alla loro morte. Il termine "ipertensione polmonare primaria" è stato ora sostituito con "ipertensione arteriosa polmonare idiopatica". I termini "primaria" e "secondaria" ipertensione polmonare non dovrebbero più essere usati. Ulteriori dettagli sono nella sezione Classificazione qui sotto.
Causa
La causa più comune dell'ipertensione polmonare è l'insufficienzacardiaca sinistra che porta all'ipertensione venosa polmonare. Questo può essere dovuto a un malfunzionamento sistolico o diastolico del ventricolo sinistro o a una disfunzione valvolare come il rigurgito mitrale o la stenosi mitrale. Di solito si manifesta come edema polmonare.
Le cause comuni dell'ipertensione arteriosa polmonare (IPA) includono l'HIV, la sclerodermia e altri disturbi autoimmuni, la cirrosi e l'ipertensione portale, l'anemia falciforme, le cardiopatie congenite, le malattie della tiroide e altre. L'uso di pillole per la perdita di peso come Fen-Phen, Aminorex, fenfluramina (Pondimin), e fentermina ha portato allo sviluppo di PAH in passato.
Patogenesi
Qualunque sia la causa iniziale, l'ipertensione polmonare comporta il restringimento dei vasi sanguigni collegati e all'interno dei polmoni. Questo rende più difficile per il cuore pompare il sangue attraverso i polmoni, proprio come è più difficile far scorrere l'acqua in un tubo stretto rispetto a uno largo. Col tempo, i vasi sanguigni colpiti diventano più rigidi e spessi, aumentando ulteriormente la pressione sanguigna all'interno dei polmoni e compromettendo il flusso sanguigno. Inoltre, l'aumentato carico di lavoro del cuore causa l'ispessimento e l'allargamento del ventricolo destro, rendendo il cuore meno capace di pompare il sangue attraverso i polmoni, causando l'insufficienza cardiaca destra. Poiché il sangue che scorre attraverso i polmoni diminuisce, il lato sinistro del cuore riceve meno sangue. Questo sangue può anche trasportare meno ossigeno del normale. Pertanto, diventa sempre più difficile per il lato sinistro del cuore pompare per fornire sufficiente ossigeno al resto del corpo, soprattutto durante l'attività fisica.
Diagnosi
Poiché l'ipertensione polmonare può essere di 5 tipi principali, una serie di test deve essere eseguita per distinguere l'ipertensione arteriosa polmonare dalle varietà venose, ipossiche, tromboemboliche o varie.
Viene eseguito un esame fisico per cercare i segni tipici dell'ipertensione polmonare. Questi includono suoni cardiaci alterati, come un S2 o secondo suono cardiaco ampiamente diviso, un forte suono P2 o di chiusura della valvola polmonare (parte del secondo suono cardiaco), un'onda (para)sternale, un possibile S3 o terzo suono cardiaco e un rigurgito polmonare. Altri segni includono la distensione venosa giugulare (allargamento delle vene giugulari), l'edema periferico (gonfiore delle caviglie e dei piedi), l'ascite (gonfiore addominale dovuto all'accumulo di liquidi), il reflusso epato-giugulare e il clubbing.
Ulteriori procedure sono necessarie per confermare la presenza di ipertensione polmonare ed escludere altre possibili diagnosi. Queste generalmente includono test di funzionalità polmonare, esami del sangue, elettrocardiografia (ECG), misurazioni dei gas sanguigni arteriosi, radiografie del torace (seguite da una TAC ad alta risoluzione se si sospetta una malattia polmonare interstiziale), e scansione ventilazione-perfusione o V/Q per escludere un'ipertensione polmonare tromboembolica cronica. La biopsia del polmone di solito non è indicata a meno che l'ipertensione polmonare non sia dovuta a una sottostante malattia polmonare interstiziale. Ma le biopsie polmonari sono gravide di rischi di sanguinamento a causa dell'alta pressione sanguigna intrapolmonare. Il miglioramento clinico è spesso misurato da un "six-minute walk test", cioè la distanza che il paziente può percorrere in sei minuti. La stabilità e il miglioramento di questa misurazione sono correlati a una migliore sopravvivenza.
Anche se la pressione arteriosa polmonare può essere stimata sulla base dell'ecocardiografia, il campionamento della pressione con un catetere di Swan-Ganz fornisce la misura più precisa. La PAOP e la PVR non possono essere misurate direttamente con l'ecocardiografia. Pertanto, la diagnosi di PAH richiede un cateterismo cardiaco. Un catetere di Swan-Ganz può anche misurare la portata cardiaca, che è molto più importante nella misurazione della gravità della malattia rispetto alla pressione arteriosa polmonare.
La pressione arteriosa polmonare normale in una persona che vive a livello del mare ha un valore medio di 12-16 mm Hg (1600-2100 Pa). L'ipertensione polmonare definitiva è presente quando la pressione media a riposo supera i 25 mm Hg (3300 Pa). Se la pressione media dell'arteria polmonare sale oltre 30 mm Hg (4000 Pa) con l'esercizio, anche questo è considerato ipertensione polmonare.
La diagnosi di PAH richiede la presenza di ipertensione polmonare con altre due condizioni. La pressione di occlusione dell'arteria polmonare (PAOP o PCWP) deve essere inferiore a 15 mm Hg (2000 Pa) e la resistenza vascolare polmonare (PVR) deve essere superiore a 3 unità di Wood (240 dyn-s-cm-5 o 2,4 mN-s-cm-5).
Classificazione
Classificazione attuale
Nel 2003, il 3° Simposio Mondiale sull'Ipertensione Arteriosa Polmonare è stato convocato a Venezia per modificare la classificazione in base alla nuova comprensione dei meccanismi della malattia. Il sistema rivisto sviluppato da questo gruppo fornisce il quadro attuale per la comprensione dell'ipertensione polmonare.
Il sistema include diversi miglioramenti rispetto al precedente sistema di classificazione di Evian del 1998. Le descrizioni dei fattori di rischio sono state aggiornate e la classificazione degli shunt sistemico-polmonari congeniti è stata rivista. Una nuova classificazione dei fattori genetici nel PH è stata raccomandata, ma non implementata perché i dati disponibili sono stati giudicati inadeguati.
Il sistema di classificazione rivisto di Venezia 2003 può essere riassunto come segue:
- WHO Gruppo I - Ipertensione polmonare arteriosa (PAH)
- WHO Gruppo II - Ipertensione polmonare associata a malattia del cuore sinistro
- Gruppo WHO III - Ipertensione polmonare associata a malattie polmonari e/o ipossiemia
- Gruppo OMS IV - Ipertensione polmonare dovuta a malattia cronica trombotica e/o embolica
- OMS Gruppo V - Varie
Terminologia precedente
I termini ipertensione polmonare primaria e secondaria (PPH e SPH) sono stati usati in passato per classificare la malattia. Questo portava a pensare che solo la malattia primaria dovesse essere trattata, e che la varietà secondaria dovesse essere ignorata in favore del trattamento della sola malattia di base. In realtà tutte le forme di ipertensione arteriosa polmonare sono trattabili. Purtroppo, questo sistema di classificazione persiste ancora nella mente di molti medici, e probabilmente porta a molti pazienti a cui viene negato il trattamento. Questo approccio nichilista all'ipertensione arteriosa polmonare può anche contribuire alla sottodiagnosi. Si stima che ci siano circa 100.000 pazienti con PAH negli Stati Uniti, ma solo 15-20.000 sono stati diagnosticati. Molti altri sono stati erroneamente diagnosticati come BPCO, asma o insufficienza cardiaca congestizia.
Il termine ipertensione polmonare primaria (PPH) è stato sostituito con ipertensione arteriosa polmonare idiopatica (IPAH) in gran parte della letteratura medica. Tuttavia, alcuni medici continuano a usare la vecchia classificazione in modo inappropriato.
Epidemiologia
L'IPAH è una malattia rara con un'incidenza di circa 2-3 per milione all'anno e una prevalenza di circa 15 per milione. Le donne hanno quasi tre volte più probabilità di presentare l'IPAH rispetto agli uomini.
Altre forme di IPA sono molto più comuni. Nella sclerodermia l'incidenza è stata stimata dal 6 al 60% di tutti i pazienti, nell'artrite reumatoide fino al 21%, nel lupus eritematoso sistemico dal 4 al 14%, nell'ipertensione portale dal 2 al 5%, nell'HIV circa lo 0,5% e nella malattia falciforme dal 20 al 40%.
Le pillole dietetiche come il Fen-Phen hanno prodotto un'incidenza annuale di 25-50 per milione all'anno.
Trattamento
Il trattamento è determinato dal fatto che il PH sia arterioso, venoso, ipossico, tromboembolico o vario. Poiché l'ipertensione venosa polmonare è sinonimo di insufficienza cardiaca congestizia, il trattamento consiste nell'ottimizzare la funzione ventricolare sinistra mediante l'uso di diuretici, beta-bloccanti, ACE-inibitori, ecc. o nella riparazione/sostituzione della valvola mitrale o aortica.
Nella PAH, i cambiamenti dello stile di vita, la digossina, i diuretici, gli anticoagulanti orali e l'ossigenoterapia sono considerati una terapia convenzionale, ma non hanno mai dimostrato di essere benefici in modo randomizzato e prospettico.
I calcioantagonisti ad alte dosi sono utili solo nel 5% dei pazienti con IPAH che risultano vasoreattivi con il catetere di Swan-Ganz. Sfortunatamente, i calcio-antagonisti sono stati ampiamente abusati, essendo prescritti a molti pazienti con PAH non vasoreattiva, portando a un eccesso di morbilità e mortalità.
Sostanze vasoattive
Tre vie principali sono coinvolte nella proliferazione e contrazione anormale delle cellule muscolari lisce dell'arteria polmonare nei pazienti con ipertensione arteriosa polmonare. Queste vie corrispondono a importanti obiettivi terapeutici in questa condizione e giocano un ruolo nel determinare quale delle tre classi di farmaci antagonisti del recettore dell'endotelina, inibitori della fosfodiesterasi tipo 5 e derivati della prostaciclina saranno utilizzati.
La prostaciclina (prostaglandina I2) è comunemente considerata il trattamento più efficace per la PAH. L'epoprostenolo (prostaciclina sintetica, commercializzata come Flolan®) viene somministrato tramite infusione continua che richiede un catetere venoso centrale semipermanente. Questo sistema di somministrazione può causare sepsi e trombosi. Flolan® è instabile, e quindi deve essere tenuto nel ghiaccio durante la somministrazione. Poiché ha un'emivita da 3 a 5 minuti, l'infusione deve essere continua (24/7), e l'interruzione può essere fatale. Sono stati quindi sviluppati altri prostanoidi. Il treprostinil (Remodulin®) può essere somministrato per via endovenosa o sottocutanea, ma la forma sottocutanea può essere molto dolorosa. Iloprost (Ilomedin®) è anche usato in Europa per via endovenosa e ha un'emivita più lunga. Iloprost (commercializzato come Ventavis®) è l'unica forma inalata di prostaciclina approvata per l'uso negli Stati Uniti e in Europa. Questa forma di somministrazione ha il vantaggio della deposizione selettiva nei polmoni con meno effetti collaterali sistemici.
L'antagonista doppio (ETA e ETB) del recettore dell'endotelina bosentan (commercializzato come Tracleer®) è stato approvato nel 2001. Due antagonisti selettivi del recettore dell'endotelina (solo ETA) sono in fase finale di approvazione: sitaxsentan e ambrisentan. Il sildenafil, un inibitore selettivo della fosfodiesterasi di tipo 5 (PDE5) specifica per il cGMP, è stato approvato per il trattamento della PAH nel 2005. È commercializzato per la PAH come Revatio®. Il tadalafil (attualmente commercializzato come Cialis® per la disfunzione erettile) è attualmente in Fase III di sperimentazione. Il peptide intestinale vasoattivo per inalazione dovrebbe entrare negli studi clinici per la PAH nel 2007. PRX-08066 è un antagonista della serotonina attualmente in fase di sviluppo per l'ipertensione polmonare ipossica.
Chirurgia
La settostomia atriale è una procedura chirurgica che crea una comunicazione tra gli atri destro e sinistro. Allevia la pressione sul lato destro del cuore, ma al costo di livelli più bassi di ossigeno nel sangue (ipossia). È meglio eseguirlo in centri esperti. Il trapianto di polmone cura l'ipertensione arteriosa polmonare, ma lascia al paziente le complicazioni del trapianto e una sopravvivenza di circa 5 anni.
La tromboendarterectomia polmonare (PTE) è una procedura chirurgica utilizzata per l'ipertensione polmonare tromboembolica cronica. È la rimozione chirurgica di un trombo organizzato (coagulo) insieme al rivestimento dell'arteria polmonare; è una procedura ampia e molto difficile che viene attualmente eseguita in pochi centri selezionati. Le serie di casi mostrano un notevole successo nella maggior parte dei pazienti.
Il trattamento per le varietà ipossiche e varie di ipertensione polmonare non è stato stabilito. Tuttavia, studi su diversi agenti stanno attualmente arruolando pazienti. Molti medici tratteranno queste malattie con gli stessi farmaci della PAH, finché non saranno disponibili opzioni migliori.
Prognosi
Il registro NIH IPAH degli anni '80 ha mostrato una sopravvivenza mediana non trattata di 2-3 anni dal momento della diagnosi, con la causa di morte che di solito è l'insufficienza ventricolare destra (cor pulmonale). Sebbene questa cifra sia ampiamente citata, oggi è probabilmente irrilevante. I risultati sono cambiati drasticamente negli ultimi due decenni. Questo può essere dovuto a una terapia farmacologica più recente, a una migliore assistenza generale e a una diagnosi più precoce (lead time bias). Un recente studio sui risultati dei pazienti che avevano iniziato il trattamento con bosentan (Tracleer®) ha mostrato che l'86% dei pazienti era vivo a 3 anni. Con agenti multipli ora disponibili, la terapia combinata è sempre più utilizzata. L'impatto di questi agenti sulla sopravvivenza non è noto, poiché molti di essi sono stati sviluppati solo di recente. Non sarebbe irragionevole aspettarsi che la sopravvivenza mediana superi i 10 anni nel prossimo futuro.
Domande e risposte
D: Che cos'è l'ipertensione polmonare o PH?
R: L'ipertensione polmonare o PH è una condizione in cui la pressione sanguigna nei polmoni è elevata.
D: Quali sono i sintomi dell'ipertensione polmonare?
R: I sintomi dell'ipertensione polmonare comprendono difficoltà di respirazione, vertigini, affaticamento e svenimento.
D: Perché alcune persone con ipertensione polmonare hanno bisogno di ossigeno supplementare?
R: Alcune persone con ipertensione polmonare hanno bisogno di ossigeno supplementare perché la condizione rende difficile la respirazione.
D: Quando peggiorano i sintomi dell'ipertensione polmonare?
R: I sintomi dell'ipertensione polmonare peggiorano quando si fa esercizio fisico o si lavora intensamente.
D: Perché l'ipertensione polmonare è una condizione grave?
R: L'ipertensione polmonare è una condizione grave perché rende più difficile al cuore pompare il sangue e può essere fatale.
D: Qual è il nome completo dell'ipertensione polmonare?
R: Il nome completo dell'ipertensione polmonare è ipertensione arteriosa polmonare, anche se la maggior parte delle persone la chiama pah, ph o pha.
D: Di cosa possono aver bisogno alcune persone molto malate con ipertensione polmonare per vivere?
R: Alcune persone molto malate con ipertensione polmonare possono avere bisogno di un trapianto di polmoni o di un trapianto cuore-polmoni per vivere.
Articoli correlati
Autore
AlegsaOnline.com Ipertensione polmonare Leandro Alegsa
URL: https://it.alegsaonline.com/art/80022
Fonti
- ncbi.nlm.nih.gov : PMID 8692238
- ncbi.nlm.nih.gov : PMID 14985486
- ncbi.nlm.nih.gov : PMID 10555089
- ncbi.nlm.nih.gov : PMID 13679525
- ncbi.nlm.nih.gov : PMID 10903931
- ncbi.nlm.nih.gov : PMID 14659797
- ncbi.nlm.nih.gov : PMID 15194171